Фармаконадзор – это вид деятельности, связанный с определением, сбором, оценкой, изучением и предупреждением возникновения побочных реакций (далее – ПР) или других вопросов, связанных с безопасностью и эффективностью применения лекарственных средств.
Порядок ведения фармаконадзора в Украине регламентирован Приказом МЗ Украины от 27.12.2006 г. №898 «Порядок осуществления фармаконадзора» (в редакции Приказа МЗ Украины от 26.09.2016 №996), который гармонизирован с международными стандартами, включая Директиву ЕС 2001/83 и Постановление Совета ЕС 2309/93.
Обязательство по ведению фармаконадзора применимо ко всем зарегистрированным в Украине лекарственным средствам с момента их государственной регистрации. Ответственность за создание и поддержание системы фармаконадзора возложена на Заявителя/ владельца регистрационного свидетельства. Заявитель обязан назначить УЛОФ – уполномоченное лицо, ответственное за фармаконадзор в Украине. Такой человек должен иметь высшее медицинское/ фармацевтическое образование (провизор, клинический провизор) и необходимую подготовку.
Осуществление фармаконадзора за ПР лекарственных средств со стороны государства выполняет Государственный Экспертный Центр МЗ Украины (далее – ГЭЦ). В свою очередь, ГЭЦ организовывает работу врачей всех медицинских учреждений всех форм собственности, и работу всех Заявителей/ владельцев регистрационных свидетельств на разрешенные к применению в Украине лекарственные средства.
Деятельность по фармаконадзору можно разделить на два уровня:
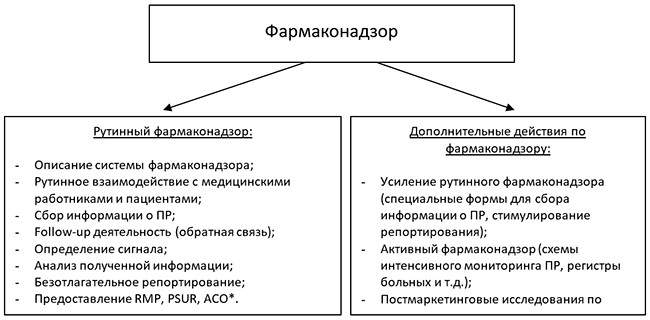
* RMP – план управления рисками (англ. Risk Management Plan), PSUR – периодически обновляемый отчет по безопасности (англ. Periodic Safety Update Report), ACO – Дополнение к обзору клинических данных (англ. Addendum to the Clinical Overview).
Рутинный фармаконадзор должен быть организован Заявителем/ владельцем регистрационного свидетельства. Рутинный фармаконадзор включает в себя следующие действия со стороны Заявителя/ или представителя Заявителя:
- Описание системы фармаконадзора: наличие, контактные данные и биографическая справка об УЛОФ, информация о мастер-файле, гарантийное письмо Заявителя о наличии необходимых ресурсов для выполнения фармаконадзора в Украине;
- Получение оперативной информации от ГЭЦ касательно всех серьезных побочных явлений (далее – ПЯ) лекарственного средства (инвалидность, смерть и прочие серьезные ПЯ). Данная информация может привести к инициированию мероприятий по минимизации выявленного риска (например, внесение изменений в инструкцию по медицинскому применению), поэтому, в соответствии с международным законодательством, должна быть распространена среди регуляторных органов других стран;
- Получение ретроспективной информации от ГЭЦ для формирования регистрационного досье для регистрации/перерегистрации ЛС в других странах;
- Предоставление оперативной информации в ГЭЦ относительно всех серьезных ПР лекарственного средства, которые были зафиксированы в Украине (например, информация от медицинских представителей, прямые обращения докторов или пациентов, информация о ПЯ из других источников);
- Предоставление оперативной информации в ГЭЦ относительно всех серьезных ПЯ, которые привели к смерти или угрозе жизни пациентов на территории других стран;
- Предоставление оперативной информации в ГЭЦ относительно отсутствия эффективности лекарственного средства при лечении жизненно небезопасных состояний, неотложных состояний и состояний, при которых отсутствие эффекта может угрожать жизни пациента;
- Предоставление оперативной сводной информации (включая развернутую информацию, методы коррекции ПЯ) обо всех побочных явлениях и/или обо всех случаях отсутствия эффективности лекарственного средства на запрос ГЭЦ;
- Подготовка и подача консолидированной информации о состоянии безопасности медицинского применения лекарственного средства в Украине для перерегистрации (подготовка «локального» PSUR);
- Участие в прочих аспектах сотрудничества с ГЭЦ и всеми остальными субъектами фармаконадзора;
- Периодическая подача PSUR. Начиная с момента международной даты рождения лекарственного средства, и после его регистрации в Украине, необходимо подавать PSUR с такой периодичностью:
Для впервые зарегистрированных ЛС:
- 1 раз в 6 мес (2 года),
- 1 раз в год (2 года),
- 1 раз в 3 года (начиная с даты регистрации)
Далее:
- в соответствии со сроками ЕМА или
- в соответствии со сроками в регистрационном свидетельстве,
- по требованию регуляторного органа.
Подача RMP:
- при регистрации/перерегистрации ЛС;
- в случае изменений, требующих новой регистрации (новая лекарственная форма, новый способ введения, новый процесс производства для биотехнологических ЛС, педиатрические показания, существенные изменения показаний);
- в случае появления/выявления новых рисков (изменение спецификации, плана по фармаконадзору, мер по минимизации рисков, соотношения польза/риск);
- по требованию регуляторного органа.
Подача АСО – при перерегистрации ЛС.
Существуют определенные требования к форме подачи PSUR, RMP, АСО: документы должны иметь установленную структуру, определенные разделы должны подаваться в переводе на украинский язык, они должны быть сопровождены письмами и формулярами установленного формата (сравнительное описание трех документов.PDF).
Компания «Кратиа» готова предложить услуги по выполнению отдельных частей или ведению всей системы фармаконадзора Вашей компании в Украине. Компания «Кратиа» располагает специалистами соответствующего профиля, которые обладают необходимыми знаниями, опытом и навыками. Мы готовы предложить:
- полное выполнение функций по созданию и ведению фармаконадзора, назначение нашего сотрудника ответственным за фармаконадзор лекарственных средств Вашей компании;
- мониторинг, анализ, обработка и подача сообщений о серьезных ПЯ из других стран (CIOMS или MedWatch);
- обучение Вашего сотрудника, написание СОП по фармаконадзору;
- проведение тренингов для сотрудников Вашей компании;
- мониторинг СМИ по выбранным ЛС;
- разработку (написание) или доработку Плана управления рисками (Risk Management Plan – RMP), Периодически обновляемого отчета по безопасности (Periodic Safety Update Report – PSUR), Дополнения к обзору клинических данных (Addendum to the Clinical Overview – ACO).











